Click here to see other posts about XPS
Only 10 $ for interpretation of each element in your XPS spectrum Payment Upon Completion Send your spectra...
X-Ray photoelectron spectroscopy (XPS), also known as electron spectroscopy for chemical analysis (ESCA) is a non-destructive technique used to analyze the surface of a material. The XPS will measure the elemental composition, chemical state as well as the electronic state, thickness measurements of overlayers (up to 8nm), and will give you the empirical formula of the material that is being analyzed. This instrument will only detect elements with an atomic number higher of 3 and higher since hydrogen and helium atoms are very small and the probability of detecting them is almost zero. Also, it can only analyze depths ranging from 1 to 10nm, for this reason it only gives analysis of the surface. Preparation of the samples is minimal if any; you can analyze samples “as receive” or can clean the surface to eliminate any contaminates that might be present. Some examples that can be analyzed using the XPS are elements, metal alloys, semiconductors polymers, ceramics, and inorganic compounds. Other examples include paints, inks, viscous oils, wood and papers.

Physics of XPS

The XPS functions by irradiating a surface with a beam of x-rays which are usually monochromatic Al Ka (1486.6eV) or non-monochromatic Mg Ka (1253.6eV) in an ultra-high vacuum. When the x-ray photons hit the sample, they transfer this energy to core electrons and are emitted from the initial state with a kinetic energy which is being measured (Figure 1). It will also count the number of photoelectrons that are being ejected from the surface with the cylindrical mirror detector analyzer. With this information you can obtain an XPS spectrum which plots the number of electrons detected vs. the binding energy of the electrons detected (Figure 2). Since each element will produce a characteristic peak at characteristic binding energies, the element at the surface can be identified and because the number of electrons in each peak is directly related to the amount of the element, the elemental composition within the area that is being analyzed can be calculated. There are tables with the kinetic energies as well as binding energies already in the system that will help identify the elements present in the surface of the material.
The binding energy of each emitted electron can be calculated using the equation below since the energy of the x-rays being emitted is known.
Ebinding = Ephoton– (Ekinetic + F)
Ebinding is the binding energy of the electron, Ephoton is the energy of the x-ray photons, Ekinetic is the kinetic energy that is measured by the XPS, and F is the work function of the spectrometer.
Figure 1. XPS, sample is being irradiated by x-rays which will then emit core electrons which are then detected and data is collected to obtain a spectrum.

References
- X-Ray Photoelectron Spectroscopy, In National Physical Laboratory, Retrieved October 29, 2012 form http://www.npl.co.uk/science-technology/surface-and-nanoanalysis/surface-and-nanoanalysis-basics/introduction-to-xps-x-ray-photoelectron-spectroscopy
- XPS Works, Actinide Research Quarterly, Retrieved October 29, 2012 form http://arq.lanl.gov/source/orgs/nmt/nmtdo/AQarchive/04summer/XPS.html
- X-Ray Photoelectron Spectroscopy, In Wikipedia, Retrieved October 29, 2012 from http://en.wikipedia.org/wiki/X-ray_photoelectron_spectroscopy
Torres, D., X-Ray Photoelectron Spectroscopy (XPS),The University of Texas at El Paso, Retrieved October 29, 2012
Example Of XPS
XPS depth profiling study on the passive oxide film of carbon steel in saturated calcium hydroxide solution and the effect of the chloride on the film properties
By: P.Ghods, O.B. Isgor, J.R. Brown, F. Benseabaa, D. Kingston
Introduction
The purpose of the paper presented was to use XPS in order to characterize the passive oxide layer that forms on carbon steel rebar in concrete pore solutions when it is passivated in calcium hydroxide solutions. Since there is very few information on the compositional characteristics of the passive oxide film before and after it has been exposed to this high alkaline environments, they decided to use XPS since it will give the depth profiling of the surface.
Preparation of Specimens
An analysis was conducted on the cross-sections of four carbon steel rebar specimens, which were 8-mm long each. The size designation for the rebar was #10M. The specimens where then hot-mounted and polished to 0.05µm and used isopropyl alcohol in order to prevent oxidation. The epoxy was then removed and three samples were submerged into saturate calcium hydroxide (CH) solution (99.6% CH in distilled water). The first sample (CH-2) was taken out after 2 days and the second one, CH-9, after 9 days. The third sample (CH-Cl) after 9 days, was then submerged in to a 0.05M chloride solution for 14 more days. This was done in order for the chlorides to react with the passivation film. The three specimens after they were taken out of the solution were placed in a jar containing isopropyl alcohol until the use of XPS was needed. The final specimen, AE, was exposed to indoor air at room temperature for 10 minutes to allow oxidation of the steel surface.
XPS Procedures
In this experiment, they used a PerkinElmer PHI-5700-2 XPS spectrometer that used an achromatic Al Ka x-ray source. It contained an electronic ultra-high vacuum chamber with pressure of 10-6 Pa and was operated at 15kV. The work function was calibrated using ultra-pure gold metal. The information was obtained by using a spherical capacitor analyzer, which was at an angle of 54? with the x-ray source, and the x-ray source was at an angle of 90? with the specimen surface. The analyzed area was 800µm.
In order to collect low-energy spectra, they did a survey scan which had the following conditions: energy range=1400eV, analyzer pass energy= 187.8eV, step size= 0.25 and a sweep time of 180s. In order to obtain high-resolution spectra, they used 10 or 20eV spectral windows at an analyzer pass energy of 29.3eV and 0.1eV steps. The spectra was for oxygen (O 1s), carbon (C1s), iron (Fe 2p), chlorine (Cl 2P), calcium (Ca 2p), and sodium (Na 1s). They collected and processed all survey and high-resolution spectra using PHI Access XPS operating software.
XPS Data Analysis

To figure out the sampling depth (d) which is the thickness of the layer, they used the equation below,
d= 3?cos?
where ? is the decreasing length and ? is the take -off angle with respect to the surface normal, which would be zero for this case. To find the kinetic energy the equation below was used,
Ek=hv- Eb
Where Ek is the kinetic energy, h is Planck’s constant, v is frequency, and Eb is the binding energy. Table 2 below shows the calculation of the sampling depth for iron oxide. The average sampling depth was of 8.5nm.
Curve fitting had to be done to the high-resolution spectra in order to get the minimum number of peaks that will result in an optimum fit. This was done using Casa XPS software and setting a few constraints in order to get the optimal fitting. Constrains included, setting peak positions to the average reported data in literature, peak positions were set constant for all depths, the full width at half maximum (FHWM) were set to the FHWM of the photoelectron core level of each element, Sheirley background corrections algorithms were used, peaks were calibrated to hydrocarbon signal set at 285eV, and semi-quantitative composition data was collected by using XPS elemental sensitivity factors. The curves that were fitted were for Fe 2p, O 1s, C 1s, and Ca 2p spectra. The curve parameters used are shown below in Table 3.

Results and Discussions
The XPS depth profiles for all four elements and for the four specimens analyzed are shown in Fig. 5 below. The graph for iron shows that as depth increases, so does the concentration, as for the oxygen curve, there will be an increase in concentration and around 2.5nm in depth it will decrease. In the carbon curves, the concentration is high at the surface which is most likely due to contamination during preparation, but then remains constant throughout the rest of the analysis. For the calcium curves, most of the samples had a constant concentration as the depth increased except for the sample which was only exposed to air which did not contain large amounts of calcium. The reason for small amounts of calcium present in the AE specimen was because there were particles embedded on the surface during polishing. As for the constant concentrations of calcium in the rest of the sample, SEM and EDS was used and showed that it was due to CH and CaCl precipitates at the surface. It was concluded that because the precipitates contained the same elements (C,O, Ca) that were analyzed by the XPS, their spectra would not be used to study the atomic structure of the oxide film. Only the Fe 2p spectrum would be used to characterize the oxide film.

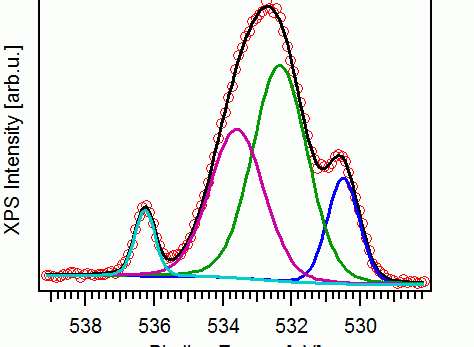
Analyzing the Fe 2p spectra at different depths for the CH-2 sample, a few observations were made. First, the five components were identified to be, iron (Fe), cementite (Fe3C), magnetite (Fe3O4), hematite/ferrihydrite (Fe2O3/ FeOOH), and Fe2O3 satellite structure; their peak position were also identified as seen in the image (Fig.8). Another observation made was that the Fe peak increased in intensity with ion sputtering which means that the Fe component comes from the substrate. Also, the sputtering shifted the Fe 2P signal to the left which indicates that the oxide film is thin no matter what the exposure time was.
The effect of exposure conditions on the thickness of the oxide film was also analyzed. Results showed that the AE specimen had a thicker iron oxide layer than the other samples and that the CH-Cl spectrum was the one with the thinnest oxide layers (See spectra below). The reason for the AE sample having a thicker layer can be due to porosity and also because the oxide layer of the CH specimens might have dissolved in the solution. The conclusion made for the CH-Cl curve was that chloride reduces the thickness of the oxide film. It was also concluded that since the spectrum for the different exposure times were almost the same, the exposure time does not affect the thickness as much. The ratio of the iron oxide to the metallic iron concentration at different sputtering depth was analyzed and results showed that above a sputtering depth of 5nm, the ratio remained constant. This meant that the iron oxide film was about 5nm in thickness. To confirm this, they used several equations and the thickness of the oxide layers to be 5.7, 4.1, 4.1, 3.6nm for AE, CH-2, CH-9, CH-Cl respectively. Other observations made where that the concentration of Fe3+ relative to Fe2+ decreased with depth in the oxide film and that longer exposure times will increase the concentration of Fe2+ relative to Fe3+.


Conclusion
In this experiment, they used the XPS depth profiling in order to characterize the oxide film of carbon steel when it was saturated in calcium hydroxide (CH) solutions and also what the effect of chloride (CH-Cl) could be on the film. Samples where carefully prepared and placed in CH and CH-Cl solutions for different amounts of time. Then they were analyzed using the XPS.
After obtaining the spectra of the specimens studied and analyzing them, they were able to make valuable conclusions. The first finding was that the carbon steel contained precipitates of calcium hydroxide and calcium carbonate so several spectra were not used for analysis. The study was only done for the Fe 2p spectrum. With the XPS depth profiles, they were able to determine the thickness of the iron oxide film to be about 4nm. The spectra for the four different specimens studies also showed that there was almost no difference between them meaning that there was no effect on exposure time to the CH solutions. Analyzing the spectra also showed that the exposure to chloride reduced the thickness of the oxide film. Another conclusion made was that there were higher concentrations of Fe2+ at the substrate and at the surface it was mostly composite of Fe3+. The longer the specimens were exposed to the CH solution, the larger the Fe2+ concentration. As seen with this experiment, the XPS is a valuable instrument that can tell us a lot about a material.
References
- Mark C. Biesinger, Brian R. Hart, Russell Polack, Brad A. Kobe, Roger St.C. Smart, Analysis of mineral surface chemistry in flotation separation using imaging XPS, Minerals Engineering, Volume 20, Issue 2, February 2007, Pages 152-162, ISSN 0892-6875, 10.1016/j.mineng.2006.08.006.
(http://www.sciencedirect.com/science/article/pii/S0892687506002093)