See more posts about the polarization test
Only 10$ per sample for interpreting of your polarization curve Payment Upon Completion Send your polarization results...
Most metallic corrosion occurs via electrochemical reactions at the interface between the metal and an electrolyte solution. For example, a thin film of moisture on a metal surface forms the electrolyte for atmospheric corrosion. A second example is when wet concrete is the electrolyte for reinforcing rod corrosion in bridges. Although most corrosion takes place in water, corrosion in non-aqueous systems is not unknown.
Corrosion normally occurs at a rate determined by an equilibrium between opposing electrochemical reactions. One reaction is the anodic reaction, in which a metal is oxidized, releasing electrons into the metal. The other is the cathodic reaction, in which a solution species (often O2 or H+) is reduced, removing electrons from the metal. When these two reactions are in equilibrium, the flow of electrons from each reaction is balanced, and no net electron flow (electrical current) occurs. The two reactions can take place on one metal or on two dissimilar metals (or metal sites) that are electrically connected.
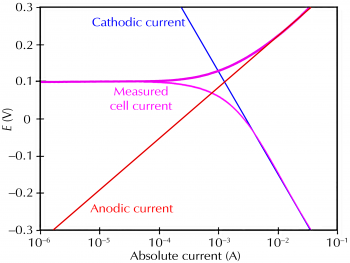
Figure 1 sketches this process. The vertical axis is electrical potential and the horizontal axis is the logarithm of absolute current. The theoretical current for the anodic and cathodic reactions is represented as straight lines. The curved line is the total current: the sum of the anodic and cathodic currents. This is the current that you measure when you sweep the potential of the metal with your potentiostat. The sharp point in the curve is actually the point where the current reverses polarity as the reaction changes from anodic to cathodic, or vice versa. The sharp point is caused by plotting along a logarithmic axis. The use of a logarithmic axis is necessary because of the wide range of current values that must be recorded during a corrosion experiment. Because of the phenomenon of passivity, the current often change by six orders of magnitude during a corrosion experiment.

Figure 1. Corrosion process showing anodic and cathodic components of current.
The potential of the metal is the means by which the anodic and cathodic reactions are kept in balance. Refer to Figure 1. Notice that the current from each half reaction depends on the electrochemical potential of the metal. Suppose that the anodic reaction releases too many electrons into the metal. Excess electrons thus shift the potential of the metal more negative, which slows the anodic reaction and speeds up the cathodic reaction. This counteracts the initial perturbation of the system.
The equilibrium potential assumed by the metal in the absence of electrical connections to the metal is called the open-circuit potential, Eoc. In most electrochemical corrosion experiments, the first step is the measurement of Eoc.
The value of either the anodic or cathodic current at Eoc is called the corrosion current, Icorr. If we could measure Icorr, we could use it to calculate the corrosion rate of the metal. Unfortunately, Icorr cannot be measured directly. However, it can be estimated using electrochemical techniques. In any real system, Icorr and corrosion rate are a function of many system parameters, including type of metal, composition of the solution, temperature, movement of the solution, metal history, and many others.
The above description of the corrosion process does not say anything about the state of the metal surface. In practice, many metals form an oxide layer on their surface as they corrode. If the oxide layer inhibits further corrosion, the metal is said to passivate. In some cases, local areas of the passive film break down, allowing significant metal corrosion to occur in a small area. This phenomenon is called pitting corrosion or simply pitting.
Because corrosion occurs via electrochemical reactions, electrochemical techniques are ideal for the study of the corrosion processes. In electrochemical studies, a metal sample with a surface area of a few square centimeters is used to model the metal in a corroding system. The metal sample is immersed in a solution typical of the metal’s environment in the system being studied. Additional electrodes are immersed in the solution, and all the electrodes are connected to a device called a potentiostat. A potentiostat allows you to change the potential of the metal sample in a controlled manner and measure the current that flows as a function of applied potential.
Both controlled-potential (potentiostatic) and controlled-current (galvanostatic) polarization are useful. When the polarization is done potentiostatically, current is measured, and when it is done galvanostatically, potential is measured. This discussion will concentrate on controlled-potential methods, which are much more common than galvanostatic methods. With the exception of open-circuit potential versus time, electrochemical noise, galvanic corrosion, and a few others, potentiostatic mode is used to perturb the equilibrium corrosion process. When the potential of a metal sample in solution is forced away from Eoc, it is referred to as polarizing the sample. The response (that is, resulting current) of the metal sample is measured as it is polarized. The response is used to develop a model of the sample’s corrosion behavior.
Quantitative Corrosion Theory
In the previous section we pointed out that Icorr cannot be measured directly. In many cases, you can estimate it from current-versus-voltage data. You can measure a logarithmic current versus potential curve over a range of about one half volt. The voltage scan is centered on Eoc. You then fit the measured data to a theoretical model of the corrosion process.
The model we use for the corrosion process assumes that the rates of both the anodic and cathodic processes are controlled by the kinetics of the electron-transfer reaction at the metal surface. This is generally the case for corrosion reactions. An electrochemical reaction under kinetic control obeys Eq. 1, the Tafel equation.
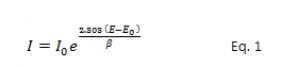 In this equation,
In this equation,
| I | is the current resulting from the reaction |
| I0 | is a reaction-dependent constant called the exchange current |
| E | is the electrode potential |
| E0 | is the equilibrium potential (constant for a given reaction) |
| β | is the reaction’s Tafel constant (constant for a given reaction, with units of volts/decade. |
The Tafel equation describes the behavior of one isolated reaction. In a corrosion system, we have two opposing reactions: anodic and cathodic.
The Tafel equations for the anodic and cathodic reactions in a corrosion system can be combined to generate the Butler-Volmer equation (Eq. 2).

where
| I | is the measured current from the cell in ampères |
| Icorr | is the corrosion current in ampères |
| E | is the electrode potential |
| Ecorr | is the corrosion potential in volts |
| βa | is the anodic β Tafel constant in volts/decade |
| βc | is the cathodic β Tafel constant in volts/decade |
What does Eq. 2 predict about the current-versus-voltage curve? At Ecorr, each exponential term equals one. The cell current is therefore zero, as you would expect. Near Ecorr, both exponential terms contribute to the overall current. Finally, as the potential is driven far from Ecorrby the potentiostat, one exponential term predominates and the other term can be ignored. When this occurs, a plot of logarithmic current versus potential becomes a straight line.
A plot of log I versus E is called a Tafel plot. The Tafel plot in Figure 1 was generated directly from the Butler-Volmer equation. Notice the linear sections of the cell current curve.
In practice, many corrosion systems are kinetically controlled and thus obey Eq. 2. A curve of logarithmic current versus potential that is linear on both sides of Ecorr is indicative of kinetic control for the system being studied. However, there can be complications, such as:
- Concentration polarization, where the rate of a reaction is controlled by the rate at which reactants arrive at the metal surface. Often cathodic reactions show concentration polarization at higher currents, when diffusion of oxygen or hydrogen ion is not fast enough to sustain the kinetically controlled rate.
- Oxide formation, which may or may not lead to passivation. This process can alter the surface of the sample being tested. The original surface and the altered surface may have different values for the constants in Eq. 2.
- Other effects that alter the surface, such as preferential dissolution of one component of an alloy, can also cause problems.
- A mixed control process where more than one cathodic, or anodic, reaction occurs simultaneously may complicate the model. An example of mixed control is the simultaneous reduction of oxygen and hydrogen ion.
- Finally, potential drop as a result of cell current flowing through the resistance of your cell solution causes errors in the kinetic model. This last effect, if it is not too severe, may be correctable via IR-compensation in the potentiostat.
In most cases, complications like those listed above cause non-linearities in the Tafel plot. Use with caution the results derived from a Tafel plot without a well-defined linear region.
Classic Tafel analysis is performed by extrapolating the linear portions of a logarithmic current versus potential plot back to their intersection. See Figure 2 (which is Figure 1 reprinted with annotations that demonstrate the analysis). The value of either the anodic or the cathodic current at the intersection is Icorr. Unfortunately, many real-world corrosion systems do not provide a sufficient linear region to permit accurate extrapolation. Most modern corrosion test software., performs a more sophisticated numerical fit to the Butler-Volmer equation. The measured data are fit to Eq. 2 by adjusting the values of Ecorr, Icorr, βa, and βc. The curve-fitting method has the advantage that it does not require a fully developed linear portion of the curve.

Figure 2. Classic Tafel analysis.
Polarization Resistance
Eq. 2 can be further simplified by restricting the potential to be very near to Ecorr. Close to Ecorr, the current-versus-voltage curve approximates a straight line. The slope of this line has the units of resistance (Ω). The slope is, therefore, called the polarization resistance, Rp. An Rp value can be combined with an estimate of the β coefficients to yield an estimate of the corrosion current.
If we approximate the exponential terms in Eq. 2 with the first two terms of a power-series expansion ( ) and simplify, we get one form of the Stern-Geary equation:

In a polarization resistance experiment, you record a curve of current versus voltage as the cell voltage is swept over a small range of potential that is very near to Eoc (generally ±10 mV). A numerical fit of the curve yields a value for the polarization resistance, Rp. Polarization resistance data do not provide any information about the values for the β coefficients. Therefore, to use Eq. 3, you must provide β values. These can be obtained from a Tafel plot, or estimated from your experience with the system you are testing.
Calculation of Corrosion Rate from Corrosion Current
The numerical result obtained by fitting corrosion data to a model is generally the corrosion current. We are interested in corrosion rates in the more useful units of rate of penetration, such as millimeters per year. How is corrosion current used to generate a corrosion rate? Assume an electrolytic dissolution reaction involving a chemical species, S:
S ® Sn+ + ne–
You can relate current flow to mass via Faraday’s Law.
Q = nFM Eq. 4
where
| Q | is the charge in coulombs resulting from the reaction of species S |
| n | is the number of electrons transferred per molecule or atom of S |
| F | is Faraday’s constant = 96 485 coulombs/mole |
| M | is the number of moles of species S reacting |
A more useful form of Eq. 4 requires the concept of equivalent weight. The equivalent weight (EW) is the mass of species S that will react with one Faraday of charge. For an atomic species, EW = AW/n (where AW is the atomic weight of the species).
Recalling that M = m/AW and substituting into Eq. 4 we get:

where m is the mass of species S that has reacted.
In cases where the corrosion occurs uniformly across a metal surface, the corrosion rate can be calculated in units of distance per year. Be careful: this calculation is only valid for uniform corrosion; it dramatically underestimates the problem when localized corrosion occurs!
For a complex alloy that undergoes uniform dissolution, the equivalent weight is a weighted average of the equivalent weights of the alloy components. Mole fraction, not mass fraction, is used as the weighting factor. If the dissolution is not uniform, you may have to measure the corrosion products to calculate EW.
Conversion from a weight loss to a corrosion rate (CR) is straightforward. We need to know the density, d, and the sample area, A. Charge is given by Q = It, where t is the time in seconds and I is a current. We can substitute in the value of Faraday’s constant. Modifying Eq. 5,

where
| Icorr | The corrosion current in ampères |
| K | A constant that defines the units for the corrosion rate |
| EW | The equivalent weight in grams/equivalent |
| d | Density in g/cm3 |
| A | Sample area in cm2 |
Table 1. Corrosion Rate Constants
| Units for corrosion rate | K | Units |
| mm/year (mmpy) | 3272 | mm (A-cm-year) |
| milli-inches/year (mpy) | 1.288 × 105 | milli-inches (A-cm-year) |
IR Compensation
When you pass current between two electrodes in a conductive solution, there are always regions of different potentials in the solution. Much of the overall change in potential occurs very close to the surface of the electrodes. Here the potential gradients are largely caused by ionic concentration gradients set up near the metal surfaces. Also, there is always a potential difference (a potential drop) caused by current flow through the resistance in the bulk of the solution.
In an electrochemical experiment, the potential that you wish to control or measure is the potential of a metal specimen (called the Working Electrode) versus a Reference Electrode. We are normally not interested in the potential drops caused by solution resistances because they are negligible in typical electrolyte solutions such as 1 M H2SO4 or 5% NaCl.
Gamry Instruments potentiostats, like all modern electrochemical instruments, are three-electrode potentiostats. They measure and control the potential difference between a non-current-carrying Reference Electrode and one of the two current-carrying electrodes (the Working Electrode). The potential drop near the other current-carrying electrode (the Counter Electrode) does not matter when a three-electrode potentiostat is used.
Careful placement of the Reference Electrode can compensate for some of the IR-drop resulting from the cell current, I, flowing through the solution resistance, R. You can think of the Reference Electrode as sampling the potential somewhere along the solution resistance. The closer it is to the Working Electrode, the closer you are to measuring a potential free from IR errors. However, complete IR compensation cannot be achieved in practice through placement of the reference electrode, because of the finite physical size of the electrode. The portion of the cell resistance that remains after placing the Reference Electrode is called the uncompensated resistance, Ru.
Gamry potentiostats can use current-interrupt or positive-feedback IR compensation to dynamically correct uncompensated resistance errors. In the current-interrupt technique, the cell current is periodically turned off for a very short time. With no current flowing through the solution resistance, its IR drop disappears instantly. The potential drop at the electrode surface remains constant on a rapid time scale. The difference in potential with the current flowing and without is a measure of the uncompensated IR drop.
The potentiostat makes a current-interrupt measurement immediately after it acquires each data point. The potentiostat actually takes three potential readings: E1 before the current is turned off, and E2 and E3 while it is off (see Figure 3). Normally, the latter two are used to extrapolate the potential difference, ∆E, back to the exact moment when the current was interrupted. The timing of the interrupt depends on the cell current. The interrupt time is 40 µs on the higher current ranges. On lower current ranges, the interrupt lasts longer.

Figure 3. Current-interrupt potential versus time.
In controlled potential modes, the applied potential can be dynamically corrected for the measured IR error in one of several ways. In the simplest of these, the IR error from the previous point is applied as a correction to the applied potential. For example, if an IR free potential of 1 V is desired, and the measured IR error is 0.2 V, the potentiostat applies 1.2 V. The correction is always one point behind, for the IR error from one point is applied to correct the applied potential for the next point. In addition to this normal mode, a Gamry Instruments potentiostat offers more-complex feedback modes in which the two points on the decay curve are averaged.
By default in the controlled potential modes, the potential error measured via current-interrupt is used to correct the applied potential. In the controlled current modes, no correction is required. If IR compensation is selected, the measured IR error is subtracted from the measured potential. All reported potentials are therefore free from IR error.
For a detailed theoretical discussion of uncompensated resistance, see Keith B. Oldham, et al., Analytical Chemistry, 72 (2000), 3972 and 3981.
Current and Voltage Conventions
Current polarities in electrochemical measurements can be inconsistent. A current value of –1.2 mA can mean different things to workers in different branches of electrochemistry or in different countries or even to different potentiostats. To an analytical electrochemist it represents 1.2 mA of anodic current. To a corrosion scientist it represents 1.2 mA of cathodic current. A Gamry Instruments potentiostat in default mode follows the corrosion convention for current in which positive currents are anodic and negative currents are cathodic. For the convenience of our users around the world, Gamry Instruments potentiostats can provide the current polarity as per your preference with a simple software command.
The polarity of the potential can also be a source of confusion. In electrochemical corrosion measurement, the equilibrium potential assumed by the metal in the absence of electrical connections to the metal is called the open-circuit potential, Eoc. We use the term corrosion potential, Ecorr, for the potential in an electrochemical experiment at which no current flows, as determined by a numerical fit of current-versus-potential data. In an ideal case, the values for Eoc and Ecorr are identical. One reason the two voltages may differ is that changes have occurred to the electrode surface during the scan.
With most modern potentiostats, all potentials are specified or reported as the potential of the working electrode with respect to either the reference electrode or the open-circuit potential. The former is always labeled as “vs. Eref” and the latter as “vs. Eoc”. The equations to convert from one form of potential to the other are:
E vs. Eoc = (E vs. Eref) – Eoc
E vs. Eref = (E vs. Eoc) + Eoc
Regardless of whether potentials are versus Eref or versus Eoc, one sign convention is used. The more positive a potential, the more anodic it is. More anodic potentials accelerate oxidation at the Working Electrode. Conversely, a negative potential accelerates reduction at the Working Electrode.
Some References on Corrosion Theory and Electrochemical Corrosion Tests
DC Electrochemical Test Methods, N.G. Thompson and J.H. Payer, National Association of Corrosion Engineers. ISBN: 1-877914-63-0.
Principles and Prevention of Corrosion, Denny A. Jones, Prentice-Hall, 1996. ISBN 0-13-359993-0.
Polarization Resistance Method for Determination of Instantaneous Corrosion Rates, J.R. Scully, Corrosion, 56 (2000), 199.
Several electrochemical corrosion techniques are approved by the ASTM (American Society for Testing and Materials, 100 Barr Harbor Drive, West Conshohocken, PA 19428. They may be found in Volume 3.02 of the ASTM Standards:
G 5: Potentiostatic and Potentiodynamic Anodic Polarization Measurements
G 59: Polarization Resistance Measurements
G 61: Cyclic Polarization Measurements for Localized Corrosion Susceptibility of Iron-, Nickel-, and Cobalt-Based Alloys
G 100: Cyclic Galvanostaircase Polarization
G 106: Verification of Algorithm and Equipment for Electrochemical Impedance Measurements
G 108: Electrochemical Potentiokinetic Reactivation (EPR) for Detecting Sensitization
G 150: Electrochemical Critical Pitting Temperature Testing of Stainless Steels
Electrochemical Techniques in Corrosion Engineering, National Association of Corrosion Engineers, 1986.
Corrosion Testing and Evaluation, STP 1000, Ed. R. Baboian and S.W. Dean, American Society for Testing and Materials, West Conshohocken, PA, 1991. ISBN 0-8031-1406-0.
Electrochemical Corrosion Testing, STP 727, Ed. F. Mansfeld and U. Bertocci, American Society for Testing and Materials, West Conshohocken, PA, 1979.
Corrosion and Corrosion Control, 3rd ed., Herbert H. Uhlig, John Wiley and Sons, New York, 1985.